PremPRI Method
PremPRI predicts the effects of single mutations occurring in RNA binding proteins on the protein-RNA interactions by calculating the binding affinity changes quantitatively. The multiple linear regression scoring function of PremPRI is composed of 11 sequence- and structure-based features, and is parameterized on 248 mutations from 50 protein-RNA complexes. The predictions are based on the 3D structure of the protein-RNA complex.
The PremPRI structure optimization protocol. We used BuildModel module of FoldX software package (1,2) to produce mutant structures. Then VMD program (3) was applied to add missing heavy side-chain and hydrogen atoms to both wild-type and mutant structures using the topology file of CHARMM36 force field (4). After that we carried out a 1000-step energy minimization for each complex in the gas phase during which the harmonic restraints with a force constant of 5 kcal mol-1 Å-2 were applied on the backbone atoms of all residues. The NAMD program v2.12 (5) and the CHARMM36 force field (4) were used to perform the energy minimization. The minimized structures of wild-type and mutant protein-RNA complexes were used for the following calculations of energy features.
The PremPRI energy function is composed of 11 features and all of them have significant contribution to the quality of the model (p-value < 0.01, t-test). The importance and the description of each feature are shown below:
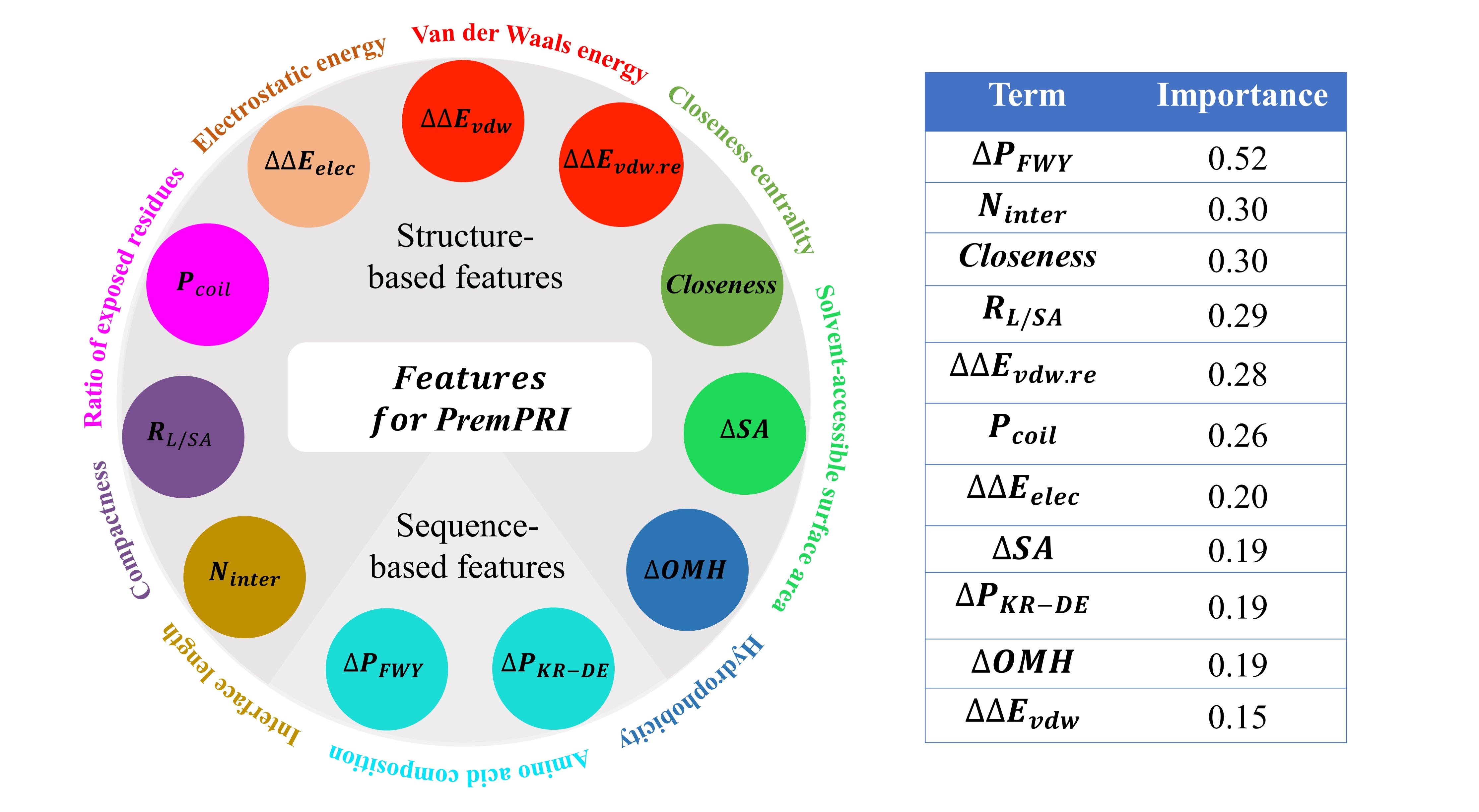
- ΔΔEvdw is the difference of Van der Waals interaction energies between mutant and wild type (ΔΔEvdw = ΔEvdwmut - ΔEvdwwt). ΔEvdw is the difference of Van der Waals energies between a protein-RNA complex and each binding partner.
- ΔΔEvdw.re is the difference of Van der Waals repulsive energies between mutant and wild type. Here, the Van der Waals repulsive energy only counts the repulsion between the residue at the mutated site and the nucleotides.
- ΔΔEelec is the difference of electrostatic interaction energies between mutant and wild type (ΔΔEelec = ΔEelecmut - ΔEelecwt). ΔEelec is the electrostatic interaction energies between the residue at the mutated site and its contact residues/nucleotides.
- Ninter is the number of amino acids at the protein-RNA binding interface. If the solvent accessible surface area of a residue in the protein is more than that in the complex, we define it as the interface residue.
- RL/SA is the ratio of protein length and its surface area. RL/SA = Length / SASA, Length and SASA is the total number of residues and the solvent accessible surface area of unbound protein, respectively.
- Closeness is the inverse of total shortest-path distance between the node of mutated site and other nodes in the remaining residue interactive network. The shortest-path distance between two nodes refers to the minimum number of nodes that reach from one node to the other (6).
- ΔSA is the difference of solvent accessible surface areas between mutant and wild type (ΔSA = SAsitemut - SAsitewt). SAsite is the solvent accessible surface area of the residue at the mutated site in the unbound protein that is extracted from the minimized complex structure.
- Pcoil = Ne.c / Nall, Ne.c and Nall are the number of exposed residues in the coil conformation and all residues in the mutated protein chain, respectively. Secondary structure elements other than α-helices and β-strands are defined as coil, which are assigned by DSSP program (7). If the ratio of the solvent accessible surface area of a residue in the complex and in solvent is more than 0.25 (8), we defined it as the exposed residue.
- ΔOMH is the difference of hydrophobicity scale between mutant and wild-type residue type. The hydrophobicity scale (OMH) for each type of amino acid residue was derived by considering the observed frequency of amino acid replacements among thousands of related structures.
- ΔPFWY and ΔPKR - DE: ΔPFWY = PFWYmut - PFWYwt, ΔPKR - DE = PKR - DEmut - PKR - DEwt, PFWY = NFWY / NAll and PKR - DE = NKR - NDE / NAll. NFWY, NKR, NDE and NAll are the number of aromatic (F, W and Y), positively charged (K and R), negatively charged (D and E) and all amino acids in the mutated protein chain, respectively.
1. Guerois, R., Nielsen, J.E. and Serrano, L. (2002) Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. Journal of molecular biology, 320, 369-387.
2. Delgado, J., Radusky, L.G., Cianferoni, D. and Serrano, L. (2019) FoldX 5.0: working with RNA, small molecules and a new graphical interface. Bioinformatics (Oxford, England), 35, 4168-4169.
3. Humphrey, W., Dalke, A. and Schulten, K. (1996) VMD: visual molecular dynamics. Journal of molecular graphics, 14, 33-38, 27-38.
4. MacKerell, A.D., Bashford, D., Bellott, M., Dunbrack, R.L., Evanseck, J.D., Field, M.J., Fischer, S., Gao, J., Guo, H., Ha, S. et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B, 102, 3586-3616.
5. Phillips, J.C., Braun, R., Wang, W., Gumbart, J., Tajkhorshid, E., Villa, E., Chipot, C., Skeel, R.D., Kale, L. and Schulten, K. (2005) Scalable molecular dynamics with NAMD. J Comput Chem, 26, 1781-1802.
6. Chakrabarty, B. and Parekh, N. (2016) NAPS: Network Analysis of Protein Structures. Nucleic acids research, 44, W375-382.
7. Joosten, R.P., te Beek, T.A., Krieger, E., Hekkelman, M.L., Hooft, R.W., Schneider, R., Sander, C. and Vriend, G. (2011) A series of PDB related databases for everyday needs. Nucleic Acids Res, 39, D411-419.
8. Brender, J.R. and Zhang, Y. (2015) Predicting the Effect of Mutations on Protein-Protein Binding Interactions through Structure-Based Interface Profiles. PLoS computational biology, 11, e1004494-e1004494.
More details can be found in our paper.

