How to use PremPLI Server?
Step 1 - Provide the structure of a protein-ligand complex
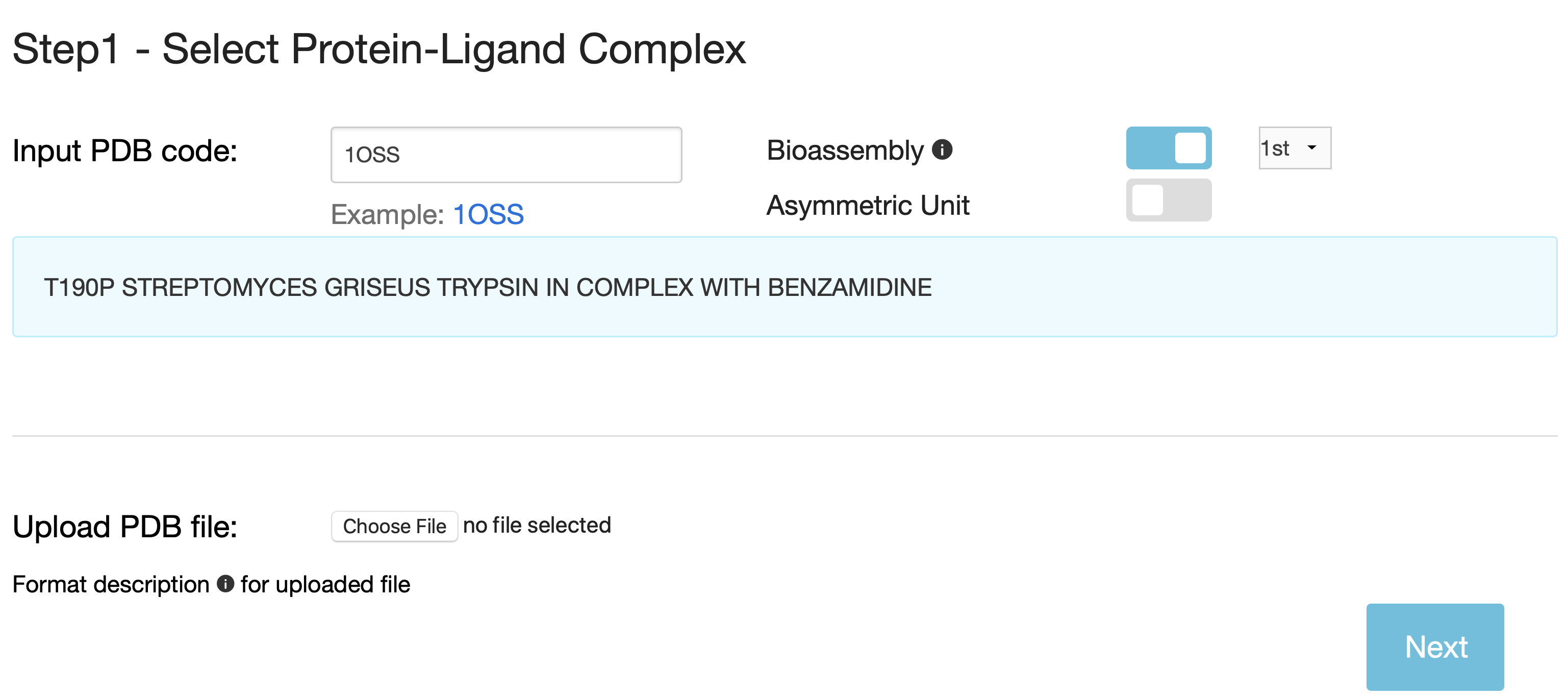
You can either input the PDB code, the structure will be retrieved from the Protein Data Bank, or upload your own structure file, which must comply with the PDB format.
In either case, the structure file must include two parts, one is protein which starts with 'ATOM', and the other is ligand which starts with 'HETATM'. Please note that:
- Bioassembly (biological unit) is the default structure. You can choose all biological assemblies for each protein-ligand complex and the first bioassembly (.pdb1) will be used as default.
- If the structure file includes multiple NMR models, only the first one will be used in calculations.
- If the uploaded structure file includes multiple protein-ligand complexes, only the first one will be considered.
- Non-protein or ligand molecules and non-standard residues or nucleotides are not taken into account during the calculations.
- PremPLI chooses the first coordinate for the atom if several different coordinates are provided in the PDB file.
Step 2 - Define two partners for a protein-ligand interaction
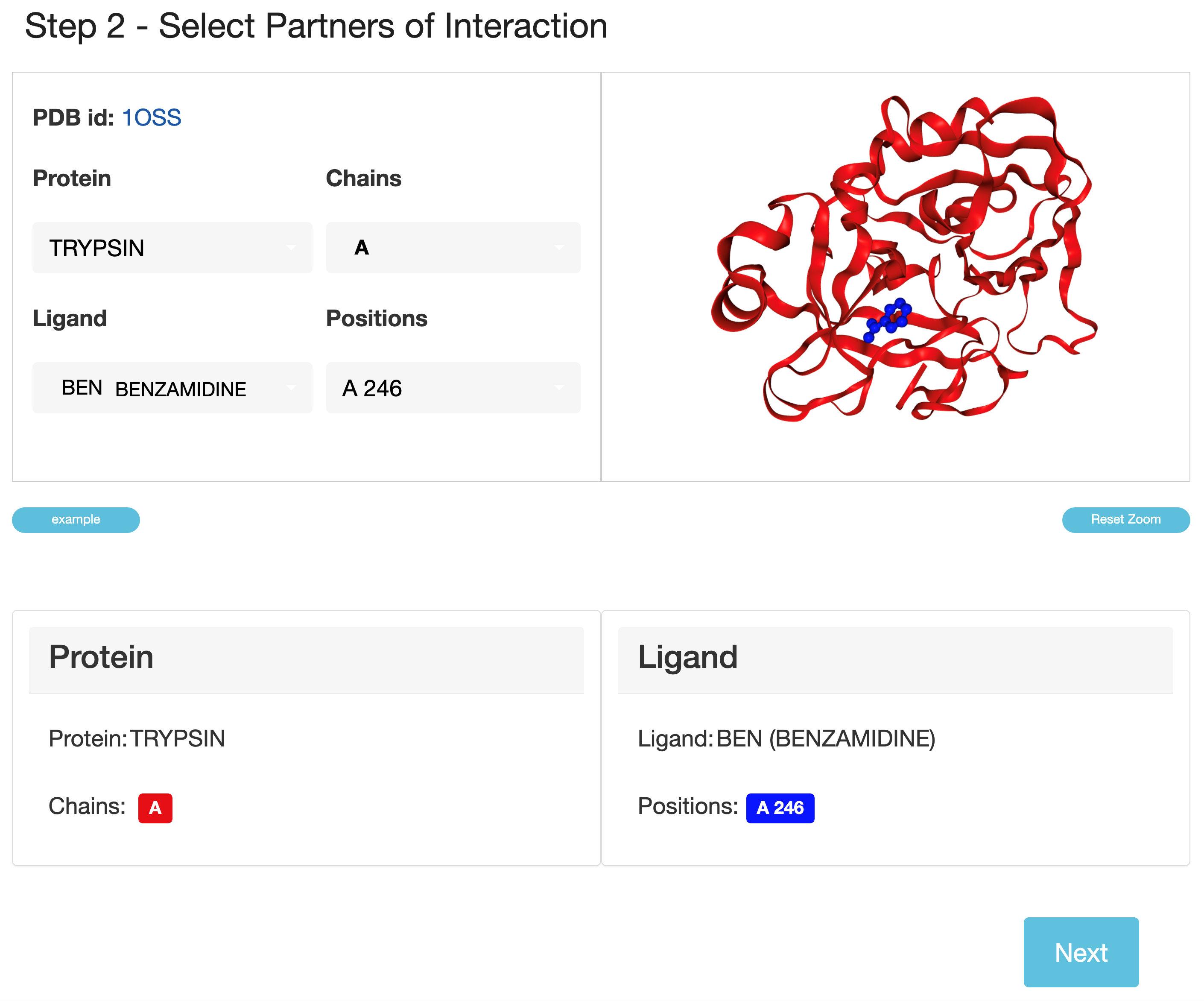
A user can define interaction partners by clicking on the button.
- PremPLI provides a 3D view of the complex colored by protein chains and ligands.
- One can assign one chain or multiple chains to Protein Chains, and one or multiple positions to Ligand Positions, but Protein Chains should include at least one chain and Ligand Positions should include at least one position; only the selected chains will be taken into account during the calculation.
- For bioassemblies if any protein or ligand/chain in the structure were generated by applying symmetry operations, they are depicted with the labels of alphanumeric combinations (for example, ‘A_1’) indicating the source molecule/chain from which they were generated.
Please note, only one type of ligand can be selected.
Step 3 - Select mutations to estimate their effects on protein-ligand interaction
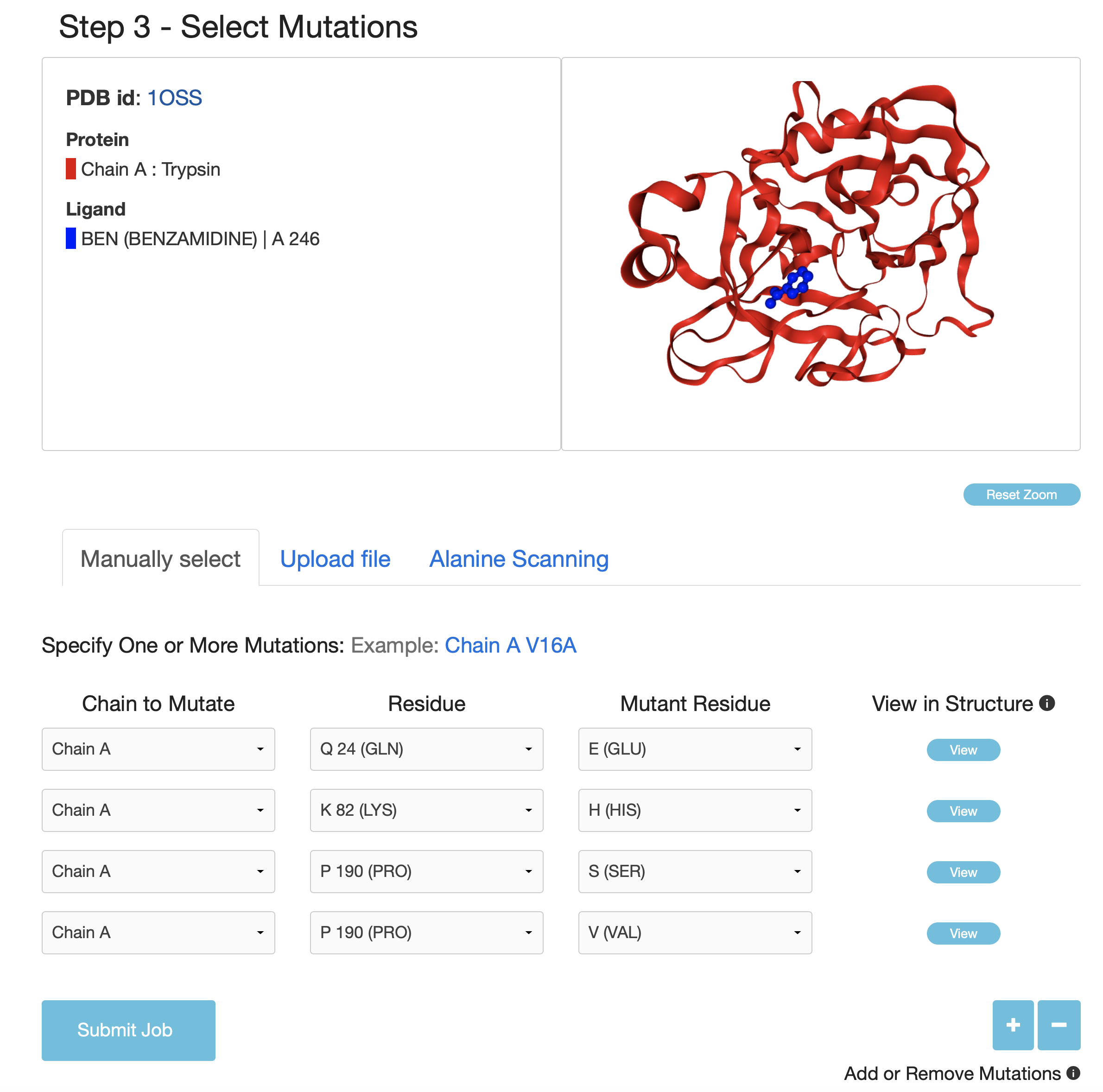
Three options are provided allowing users to do large-scale mutational scanning: "Specify One or More Mutations Manually", "Upload Mutation List" and "Alanine Scanning"
- In the option of "Specify One or More Mutations Manually", the user can submit the specified mutations and visualize each mutated site in the protein-ligand complex structure.
- "Upload Mutation List" option allows the user to upload a list of mutations in a plain text file for batch processing.
- "Alanine Scanning" option is used to perform alanine scanning for each protein chain.
Results
Upon the completion of the third step and submission of the job, PremPLI provides a job identifier for checking the job status. If an e-mail address is provided, the link to the results page will be sent via e-mail. The results will be available on this page as soon as the calculations are done. The example below shows precalculated results for job 2021031002563493343426069.
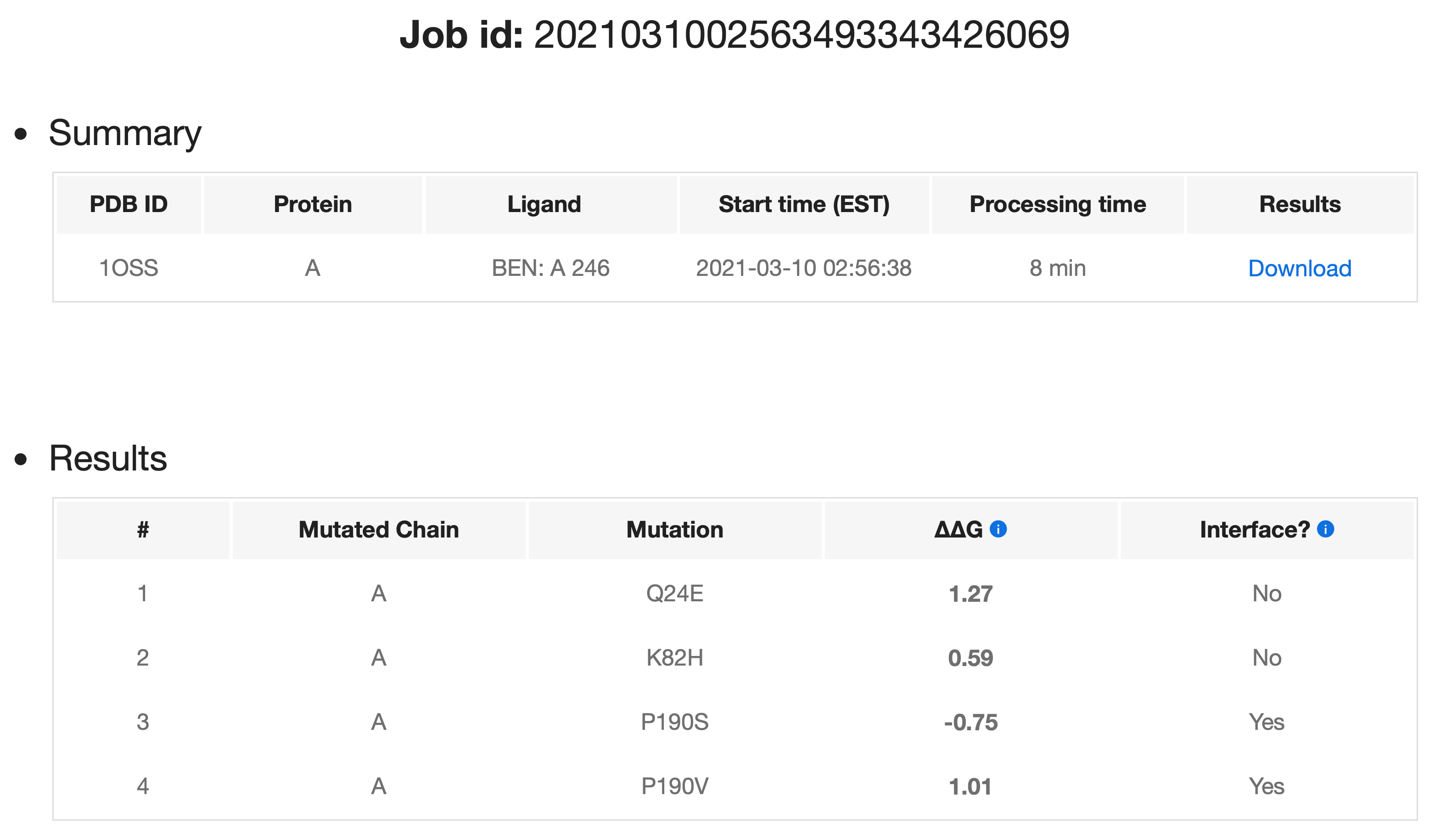
For each job, PremPLI presents a summary and actual processing time. For each single mutation in a protein-ligand complex, the PremPLI server provides:
- ΔΔG (kcal mol-1) is the predicted binding affinity change, and positive and negative signs correspond to the mutations decreasing and increasing binding affinity respectively.
- Interface (Yes/No) shows whether the mutation occurs at the protein-ligand binding interface. The interface residues were defined if any heavy atoms of them are within 5 Å distance from any heavy atoms of ligands.
Browser compatibility
| OS | Version | Chrome | FireFox | Mircosoft Edge | Safari |
| Linux | Debian10.5 | 84.0 | 86.0 | n/a | n/a |
| MacOS | 10.15.4 | 88.0 | 85.0 | n/a | 13.1 |
| Windows | 10 | 86.0 | 85.0 | 44.1 | n/a |

